

We all have heard this saying “If it’s not documented, it didn’t happen” concerning clinical trials. But the question is for clinical trials what is to be documented and how? Today we will walk through insights about source documents in clinical research.
Source means originality. Source documents are original documents, data, or records that are created during a clinical study trial. Source documents are essential documents that are required by regulatory and GCP guidelines.
There are Source Documents of 2 types:
- Electronic
- Paper
Electronic Source
Data which is audio, pictorial, text, graphics, reports, or other information created in digital form that is modified, maintained, archived, or retrieved by a computer system or electronic system.
Requirements for Electronic Source Documents:
1) Computer system (validation)
2) Electronic records (audit trail)
3) Electronic signatures
4) Users and technical support
Guidelines :
21 CFR Part 11 Guidelines; Electronic Records – Electronic signatures (1997)
Guidance for Industry: Part 11, Electronic Records; Electronic Signatures Guidelines
Scope and Application (2003)
Guidance for Industry: Computerized Systems Used in Clinical
Investigations (2007)
Electronic Source Data in Clinical Investigations – (2013) Guidance for Industry
Paper Source
Handwritten data are prepared on the pre-printed forms.
Handwritten source documents are either handwritten or printed documents (pre-filled), with the original handwritten Investigator’s signature.
The following aspects should be taken into consideration while selecting source documents:
- Design and organization of the Trial:
- Inpatient, outpatient, or combined?
- Is a site within one hospital or multicenter hospitals including remote sites.
- Who coordinates the trial (a chair of the Medical University / Academy or hospital)?
- Research documents retention (archiving) at a Medical Institution or hospital.
Documents which are considered as Source Documentation:
- Informed Consent Forms
- Medical History
- Subject Diary
- Outpatient Medical Chart
- Various Logs / Hospital Charts
- Laboratory, MRI or any other reports
- Hospital records
- Clinical and office charts
- Laboratory notes
- Subjects’ diaries or evaluation checklists
- Pharmacy dispensing records
- Recorded data from automated instruments
- Microfiches
- Photographic negatives
- Microfilm or magnetic media
- X-rays
- Subject files
- Records of pharmacy, laboratories, and other departments involved in the clinical trial
Purpose of Source documentation:
- The purpose of source documentation is to reconstruct the trial and to produce quality data.
- Source documentation is the record of the before, during, and after the trial.
- Record of accountability of the investigational product dispensed, consumed, and returned during a clinical trial.
- It is the complete medical record of the subject as the reference to the trial investigators at any point in time.
- It forms strong trial data that gets transcribed into an eCRF which is ultimately transferred into a clinical study report.
Source Document Verification (SDV)
- Source Documents Verification is a requirement of ICH GCP guidelines which CRA or monitor perform during monitoring visits;
- Site Monitoring is to verify and confirm that the created or reported trial data are accurate, original, complete, and verifiable from source documents.
The main challenges during SDV are:
- Informed Consent process
- Medical History
- Subject Diary
- Medication details
- SAE and AE reporting
- Laboratory Data
- Inappropriate ways of corrections of overwritten, missing data and loss of audit trail
- Not properly certified copies of original records
- Inclusion/exclusion criteria listed but not really confirmed
Documentation should be able to provide an audit trail to permit investigation if and when required.
Key attributes for good documentation practice described in the form of ALCOA & ALCOA+ – attributable, legible, contemporaneous, original and accurate. ALCOA+ is with few more concepts which are complete, consistent, enduring, available.
Some qualities of Good documentation practices defined by USFDA and EMEA are as follows:
- Attributable
It should be clear who has performed the action and documented the data.
Attributable

- Legible : Readable and signatures identifiable.

- Contemporaneous
The information should be documented at the correct time frame when the action performed along with the flow of events. An acceptable amount of delay should be defined and justified remark.

- Original
Original, or authorised true copy, the first record which is made by the designated person. The investigator should have the record of original source document.

- Accurate
Accurate, consistent and real representation of facts.
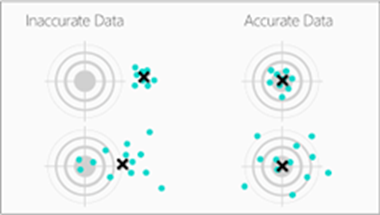
- Enduring
Long-lasting and durable.

- Available and accessible
Easily available and accessible for review or present of data during audits/inspections. The documents should be retrievable in a reasonable time.

- Complete
Complete till that point in time.

- Consistent
Demonstrate the required attributes consistently.
- Credible
Based on real and reliable facts.

- Corroborated
The data should be backed up by evidence.

Data can be captured electronically by the following ways:
- Direct entry: Data directly entered into eCRF by an authorized person (PI or Coordinator)
- Automatic transmission: Data is pulled or transferred from devices or instruments (temperature, blood pressure monitoring device, or glucometer)
- Transcription from paper or electronic source: Data is transcribed from paper to electronic medical records by an authorized person.
References:
- guideline for good clinical practice – ICH
- https://en.wikipedia.org/wiki/Source_document
- https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3121265/#:~:text=The%20most%20important%20purpose%20of,investigation%20if%20and%20when%20required.
- https://www.slideshare.net/DeepakPrabhu1/electronic-source-data-in-clinical-investigations-50383288
- https://ichgcp.net/8-essential-documents-for-the-conduct-of-a-clinical-trial
Content : Pooja Parab

